Every cell in the body contains 46 chromosomes, each of which is made up of hundreds of thousands of genes. The chromosomes are organised into 23 pairs, one chromosome of each pair coming from each parent. The two chromosomes of each pair have the same size and contain the same number of genes in both men and women for the first 22 pairs, but the 23rd pair, the “sex chromosome”, is different. For this pair, females have two equally matched “X” chromosomes, whereas males have one X-chromosome plus a few genes that only occur on the Y-chromosome. The gene for XLP1 is located at the Xq25 position on the long arm of the X-chromosome, which does not have a matching region on the Y chromosome. This gene is called “SH2DIA” (“SAP” or “DSHP”) and serves as the code for making SAP protein. Mutations in the SH2DIA gene lead to alterations in the structure and function of SAP protein or in the amount of protein produced. Although we do not know all of the functions of SAP protein, it is clear that mutations that alter the SAP protein can cause XLP1.
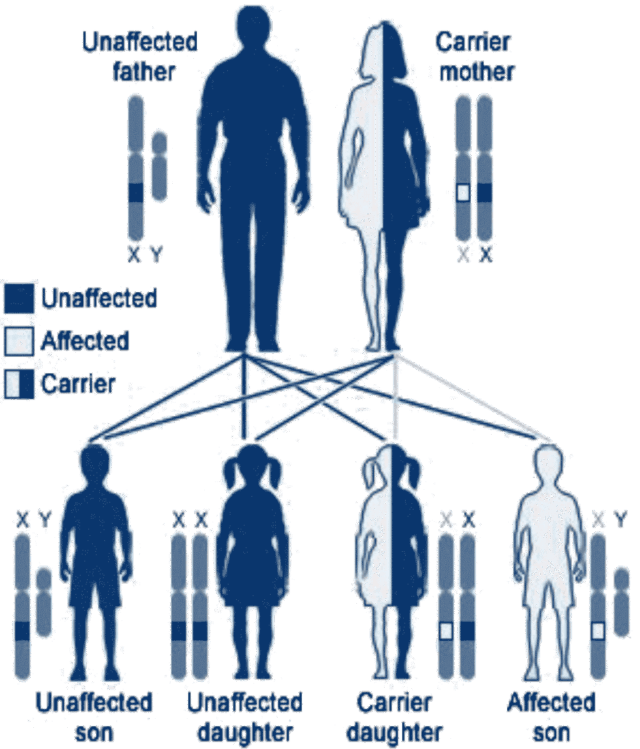
The inheritance of XLP1 follows an “X-linked” (or “sex-linked”) recessive pattern, in which females can be carriers of a SH2DIA mutation but only males will have the disease. Because females have two copies of the X-chromosome, they also have two copies of every gene on the X-chromosome. Because SH2DIA mutations are very rare, a female who inherits one chromosome carrying a mutation of the gene for XLP from one parent will almost always inherit a normal X-chromosome from the other parent. In all XLP1 mothers who carry a SH2DIA mutation on one of their two X-chromosomes, the normal SH2DIA gene on the other X chromosome provides enough SH2DIA function to prevent any disease. For this reason, a SH2DIA mutation in a female is termed a “recessive” mutation. In other words, although the female who carries a recessive SH2DIA mutation can pass the abnormal SH2DIA gene on to her children, she will not show any symptoms of XLP because of the protective effect of the normal SH2DIA gene on the other X-chromosome.
Unlike females who carry two copies of SH2DIA on their matched X-chromosomes, males have only one copy of the SH2DIA gene because they have only one X-chromosome. This is because a male who inherits from his mother an X-chromosome with one SH2DIA gene inherits from his father a shorter Y chromosome that lacks a copy of the SH2DIA gene. With no normal SH2DIA gene on the Y-chromosome to compensate for a SH2DIA mutation inherited on the mother’s X-chromosome, all males with a SH2DIA mutation will have clinical signs of XLP1.
When a mother is a carrier of a SH2DIA mutation on one of her two X-chromosomes, there is a 50% chance in any pregnancy that she will pass the X-chromosome with the SH2DIA mutation onto the child. If the child who inherits the SH2DIA mutation is a girl, she will be a carrier of XLP1, and, like her mother, be able to pass the SH2DIA mutation on to her own children. However, also like her mother, she will not show symptoms of XLP. If the child is a boy, there is a 50% chance he will inherit SH2DIA mutation and have XLP1 and a 50% chance he will inherit only the X chromosome with the normal SH2DIA gene and, therefore, not have XLP1. If the male inherits the SH2DIA mutation and has XLP,1 all of his daughters will be carriers, but none of his sons will have XLP.1 This is because the X- and Y-chromosomes are the sex-determining chromosomes, and all children who inherit his Y-chromosome (with no SH2DIA gene) from him will be boys, and all children who inherit the X-chromosome with a SH2DIA mutation will be girls.
A genetic counsellor can assist families in understanding these potential outcomes.
When a SH2DIA mutation for XLP1 cannot be detected in either parent, this is considered a new mutation or possibly a different form of XLP such as XLP2 (where the defective or missing gene is XIAP). New mutations can be sporadic or they can be a result of “gonadal mosaicism.” Gonadal mosaicism is the rare occurrence of a genetic mutation in a proportion of the eggs or sperm of an individual as a result of a random mutation that occurs during the development of eggs and sperm. The individual does not have any signs of the disorder, and the mutation would not be detected by a blood test because it is in only the egg or sperm cells. However, if one of the egg or sperm cells is used to conceive a child, that child will have the mutation in all cells.
First published May 2008.
Latest update November 2023.
This fact sheet is designed for educational purposes only and is not intended to serve as medical advice. The information provided here should not be used for diagnosing or treating a health problem or disease. It is not a substitute for professional care.
We would like to thank the Barth Syndrome Trust for their help in putting this leaflet together.